1st emergency use of COVID-19 antibody drug permitted in US




WASHINGTON — U.S. health officials have allowed emergency use of the first antibody drug to help the immune system fight COVID-19, an experimental approach against the virus that has killed more than 238,000 Americans.
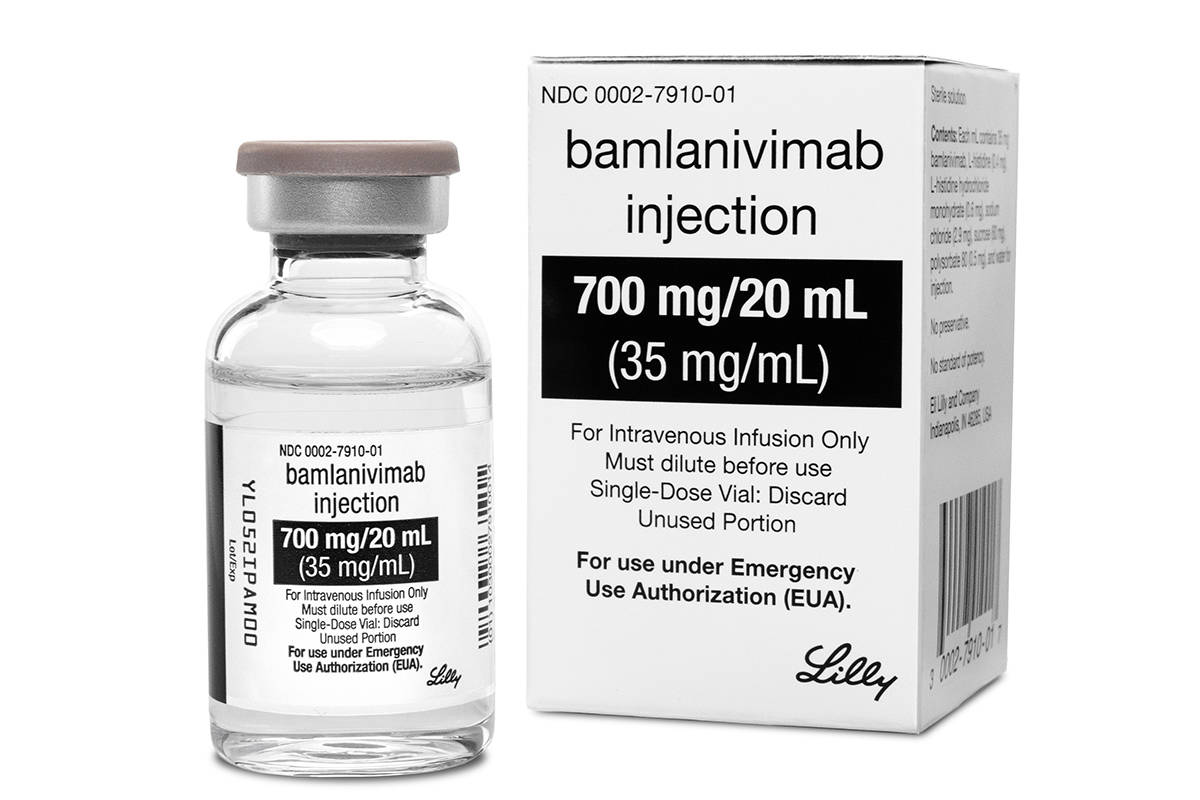
The Food and Drug Administration on Monday cleared the experimental drug from Eli Lilly for people 12 and older with mild or moderate COVID-19 not requiring hospitalization. It’s a one-time treatment given through an IV.
The therapy is still undergoing additional testing to establish its safety and effectiveness. It is similar to a treatment President Donald Trump received after contracting the virus last month.
Lilly’s studies of the antibody drug are continuing. Early results suggest it may help clear the coronavirus sooner and possibly cut hospitalizations in people with mild to moderate COVID-19. A study of it in hospitalized patients was stopped when independent monitors saw the drug did not seem to be helping in that situation.
The government previously reached an agreement to buy and supply much of the early production of Lilly’s drug.
Only one drug — Gilead Sciences’ remdesivir — has full FDA approval for treating COVID-19. Government treatment guidelines also back using dexamethasone and other steroids for certain severely ill, hospitalized patients.
One other treatment has an emergency use designation now — convalescent plasma, or the blood of COVID-19 survivors. No large studies have shown it to be more effective than usual care alone, however.
The new drug is part of an emerging family of biologic therapies that offer a promising new approach to preventing serious disease and death from COVID-19. Experts say the infused drugs could serve as a therapeutic bridge to help manage the virus until vaccines are widely available.
The drugs are laboratory-made versions of antibodies, blood proteins which the body creates to help target and eliminate foreign infections. The new therapies are concentrated versions of the antibodies that proved most effective against the virus in patient studies.
Regeneron Pharmaceuticals Inc. also has asked for emergency authorization for an antibody drug it is testing, the one Trump received.
FDA regulators authorized the Lilly drug using their emergency powers to quickly speed the availability of experimental drugs and other medical products during public health crises.
In normal times the FDA requires “substantial evidence” to show that a drug is safe and effective, usually through one or more large, rigorously controlled patient studies. But during public health emergencies the agency can lower those standards and require only that an experimental treatment’s potential benefits outweigh its risks.
The emergency authorization functions like a temporary approval for the duration of the COVID-19 pandemic. To win full approval, Lilly will have to submit additional research to fully define the drug’s safety and benefit for patients.
The government has signed an agreement with Lilly to spend $375 million to buy 300,000 vials of the drug. How many doses that would provide is unclear. Each vial contains 70 milligrams and that dose proved ineffective in the early results. It took four times that amount — 2,800 milligrams — to show any effect.
The Lilly drug is authorized for people 12 and older who weigh at least 40 kilograms (about 88 pounds), and who are at high risk for progressing to severe COVID-19 and/or hospitalization. This includes those who are 65 years of age or older, or who have certain chronic medical conditions.
AP chief medical writer Marilynn Marchione in Milwaukee contributed to this report.
The Associated Press Health and Science Department receives support from the Howard Hughes Medical Institute’s Department of Science Education. The AP is solely responsible for all content.